Dementia science has developed some remarkably sophisticated tools with the aims of understanding the course of the underlying neurodegeneration and opportunities to prevent it or slow it down. Despite valiant efforts to portray the findings in the light of a, favoured, disease model, the factual results don’t fit in. Interventions that should have worked, don’t. Among the early promises was that negligent, and therefore preventable, triggers of dementia would be identified. Traumatic brain injury and sports concussion were presumed to be among them.
The purpose of this paper is to explain: the important factual results, what really does link brain trauma and dementia and why some people develop dementia while others do not. The explanations are obvious in retrospect and provide a strong narrative basis for expert testimony in dementia liability claims.
At present, and increasingly clearly, the science indicates no causal link between either brain injury or concussion and subsequent neurodegenerative disease. Neurodegeneration is best explained as a result of biological ageing. Biological ageing occurs at different speeds in different people and at different speeds in different parts of the brain.
Dementia as an emerging liability risk
Traumatic brain injury (TBI), and concussion, are very common life events in people who now have dementia and, are very often the result of negligence. Liability for negligent brain injury as a cause of dementia is one logical result of the theoretical models and assumptions that were actively pursued between 1990 and 2015. If such causation could be proved in individual cases, the damage would be exceptionally expensive for the defendant. A high frequency high quantum long latency injury would be economically troublesome.
Has the science moved on? Yes. There is a new understanding of the relationship between TBI and dementia. That journey needs to be explained if the out-dated ideas are to be let go. Expert evidence based on the old models and assumptions can be challenged, and should be.
The brain is just the sum of its parts, or is it?
Once the mainstay of neuroscience the idea that the brain consists of distinct specialised parts with highly specific functions has been replaced by a more integrated view. Rarely, if ever, does a part act on its own. Acts in any one part depend on the state of all other parts, but to differing degrees.
At one extreme are reflexes which come close to being isolated and at the other is reason which is a highly integrative phenomenon. In the centre ground is a harmony of emotional, sensory and cognitive functions. In normal times, reason has the last word. Indeed, the concept of legal agency depends on the pre-eminence, or potential pre-eminence of reason.
Since neurones can’t be replaced, damage must be irreversible?
So long as there are enough underused neurones each part of the brain is adaptive, allowing one part to compensate for damage to itself and to another. The effect of this is also self-stabilising, such that regular usage gradually leads to improved adaptation and even full restoration of function.
Life experiences can also affect how able the brain is to adapt and compensate; highly educated people with intellectually stimulating jobs are relatively protected in this way. Childhood disadvantage and distress leave the brain less resilient.
The perpetuation of legal agency depends on the resilience of the legal person despite changes in brain biology, adaptation, knowledge and understanding. The legal agent aged 18 is the same legal agent aged 65.
Agency, integrativity and adaptation
Injury, such as post-traumatic stress disorder (PTSD), can be understood from these concept areas, as can depression.
In PTSD an initial event-related dysfunctional imbalance between emotional and cognitive powers is perpetuated, with emotion having the upper hand in some cognitive domains. An element of agency is lost, usually temporarily.
In depression an imbalance is acquired over time, often aided by misplaced agency, and then perpetuated. Agency is rarely lost, but when it is it would be right to describe it as an injury if caused by a third party.
But what about dementia? And can it be caused by a traumatic brain injury? Can it be caused by concussion? Can it be caused by heading a football?
Dementia
Dementia is the clinical manifestation of functional loss in the brain. Typical symptoms include loss of memory, language errors and passivity.
Functional loss is caused by an advancing net loss of neurones. As more neurones are lost the adaptive ability to compensate for this physical damage is eroded to the point where function changes and is eventually lost.
Different functional groupings of brain parts have different localised functional thresholds as measured by neurone loss. Some parts lose function when 10% of neurones are lost, most have a threshold at around 30% to 50% loss and one has a threshold nearer to 80% loss. However, localised loss of function can be compensated for if there is spare capacity in other brain parts. So unless a brain part is highly specialised, the effect on function is an integrative phenomenon.
Thankfully the rate of loss of neurones in most people in most brain parts is slow. Adaptation and compensation mechanisms have time to act. But eventually, unless some other event intervenes, neurone loss will mean that the given functional threshold is reached.
So what causes neurones to die?
As with all cells, neurones can be killed by mechanical trauma, chemical stress, lack of oxygen and lack of nutrients. But most often, these causes are not present in the dementia case history.
Theoreticians are also intrigued by the apparent patterns of neurodegeneration in different types of dementia. Symptoms of Parkinson’s dementia begin with disturbed sleep and loss of sense of smell for example, early Alzheimer’s may be noticed in loss of explicit experiential memory and speech fluency. While there is a very broad spectrum of symptoms and the order in which they occur, in each type of dementia there is sufficient commonality for patterns to be clinically noticed.
The natural temptation is to attach more meaning to patterns than to random events. The commonalities tend to dominate the thinking about dementia, when in fact the randomness is the dominant characteristic. Given a focus on commonalities and a belief in distinctly specialised brain parts it would be natural to think that neurone death must first occur in the early symptom-related parts of the brain. If so, then perhaps the cause of neurone death somehow spreads from there to the rest. And so began 25 years of research. One popular theory held that dying neurones release toxic components which then self-replicate, spread to other neurones, and kill them. A bit like an infection. If true then the rate of loss of neurones would initially increase exponentially but then as the uninfected neurones are used up the rate would level off.
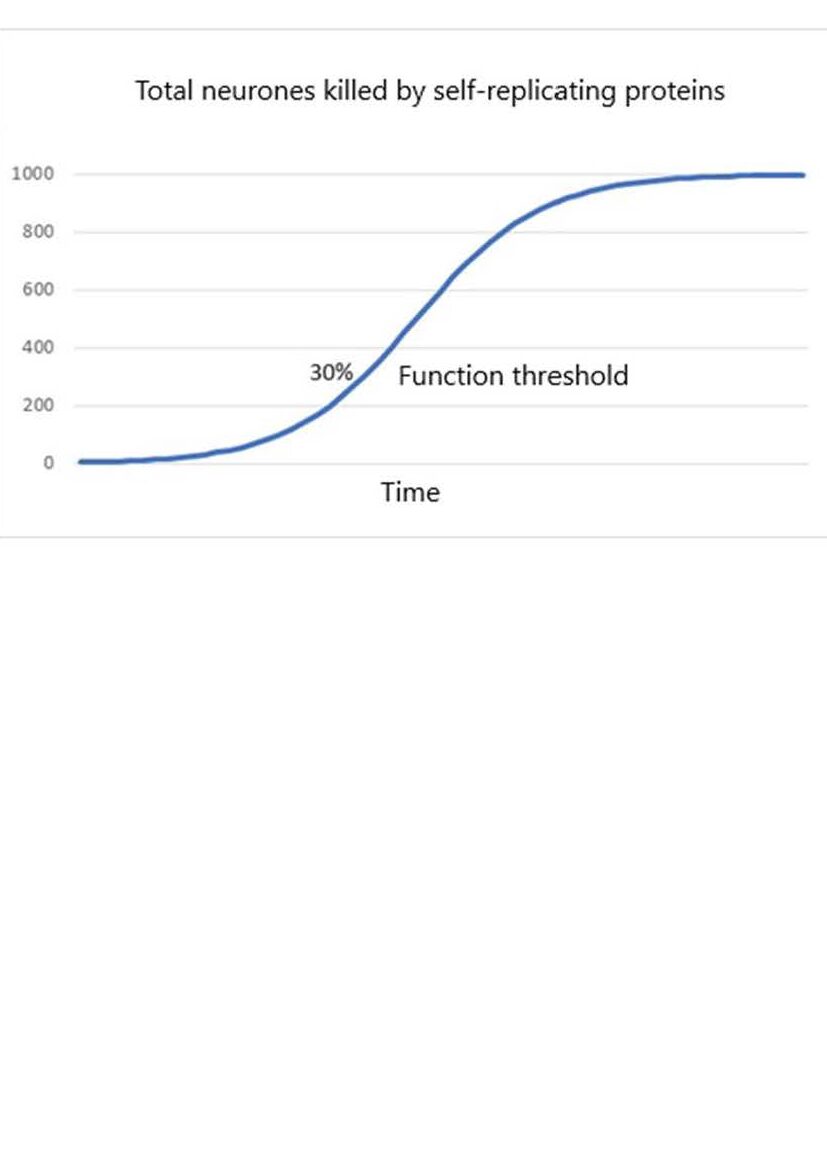
This would give rise to an overall mathematical form known as a logistic. Graphs of this kind are very common in theoretical neuroscience.
Notice that the function threshold is nearly always in a part of the logistic curve where the rate of loss of neurones is increasing.
The logistic form seems to explain why symptoms of functional loss develop more quickly as the functional threshold is approached. Occasional inexplicable changes in character and lapses of memory become more frequent as the capacity to restore normal function is used up more quickly. The explanation is logical, but as it turns out, wrong.
The identity of the proposed self-replicating neurotoxin has also evolved as techniques have improved. At one time in Alzheimer’s the culprit was the Aβ protein aggregate. It was easy to see under the microscope. Then the NFT tangle became the favourite. And then, as chemistry came forward, the pTau seed. Each being reckoned with as a logistic development.
Although often detectable before any signs of dementia, it turns out the Aβ protein aggregate has very little relationship with neurone death. Perhaps then Aβ is the cause of the deadly NFT tangle? Certainly NFT tangles are more common and increase more quickly in people with Aβ protein aggregates. However, NFT tangles begin to appear at around 10% neurone loss, so they cannot be the cause. NFT tangle density is around one seventh the density of dead neurones so most neurones die without one.
pTau seeds and NFT tangles have very similar chemistry and even though pTau seeds can be self-propagating the relationship between seed density and clinical signs is at best tenuous.
In Parkinson’s disease the culprit was α-synuclein aggregates such as Lewy bodies. These are found in many kinds of brain ageing presentation.
Yet the “infection” theory and its unstated underlying assumptions persist in some circles. One reason is that if it is true then there would be some hope that the self-propagating toxin could be intercepted by some pharmaceutic means. Medicine lives in hope of such interventions, as do patients. The biggest financial contribution to neuroscience comes from the pharmaceutical industry.
The underlying assumptions with respect to neurone death and dementia include:
- that all people’s brains are the same unless something has gone wrong and
- that all parts of any brain are the same unless something has gone wrong.
This is a very medically minded approach to logic. Understandably so, since they draw the focus on possible interventions, but it is not good science. Both assumptions must be shown to be true before building any models from them.
The theory is also attractive to those who have a need to show that the processes of dementia may be triggered by any event which causes nerve cell death. A self-propagating injury would be indivisible and 100% of the damage would be attributed to its cause.
If Aβ protein aggregates, NFT tangles and pTau seeds don’t kill neurones then what do they do? The first thing to note is that they do correlate with biological markers of neurone ageing. They can be used as markers of ageing. Cause, effect or accidental correlation is unknown. The second thing to note is that they also correspond with lower levels of energy use, as if they may reduce the metabolic activity of the cell. Evidence for this is very recent. An analogous term “silting up” may give the right image of this process.
Neurones that are less active will result in changes to the effective threshold for loss of function. That is, a person with this metabolic suppression would reach the functional threshold sooner than expected from neurone death alone.
Removal of the toxin by pharmaceutical means would restore neurone activity to some extent and where it works it would appear, for a short while, that the dementia was being cured. This has also recently been observed for the first time. There was no evidence in this very brief trial that neurone death rates were being affected.
It is too early to say for sure but the likely effect of reducing the silting up effect is to delay or temporarily reverse the clinical symptoms without making any change to the underlying process of neurone death. The benefits of this would of course be welcomed by patients if they knew it was not a cure, just a suspension of clinical progress. Such a suspension of progress would only occur in those who have metabolic interference as part of their disease.
The underlying assumptions listed as 1) and 2) above are now known to be wrong. In fact chronological age and brain age are related but differ from person to person. People with detectable levels of Aβ protein aggregates and NFT tangles have faster overall brain ageing. The obvious interpretation of the data is that every brain ages at a different rate and every part of every brain ages at a different rate. People with dementia at age 65 are those with faster brain ageing and lower resilience. Nothing has gone wrong so to speak – everyone is different.
So if there are patterns associated with the development of dementia it says only that there are subgroups of people with similar functional threshold/ ageing combinations. Within subgroups the apparent progression from one brain part to another is just a pattern in the rate of approach to threshold. The detectable effect appears to move from place to place, but the neurodegeneration does not.
It means that Parkinson’s disease and Alzheimer’s are the same at a very basic level. If they differ by the marker of ageing (e.g. Aβ vs alpha synuclein) it is because they differ in the biochemistry of those parts often first noticeably affected. In fact Aβ protein aggregates and Lewy bodies can be found in most cases of both Alzheimer’s and Parkinson’s but to different degrees and with different time profiles.
So how can you explain the clinically observed acceleration of symptoms? Surely the accelerating clinical time profile must be because neurone death rates are increasing?
Of particular note is that in all relevant studies of human brains the rate of loss of neurones follows an exponential decay law, with a higher initial rate, slowing to zero as cells are lost and finally altogether used up. Just like in radioactive decay each cell has a given probability of death in the next year and this does not depend at all (well perhaps slightly) on what has happened to other cells.
The probability of neurone death in the next year is higher for people with early dementia than in those of the same age as yet without. But the same time profile shape is found in each. Exponential decay.
But if the rate of loss is actually reducing, how come the symptom change rate increases as you get near to a formal diagnosis of dementia? The explanation can be seen graphically.
The dashed black curve shows how the capacity to maintain function decreases with time as neurones die.
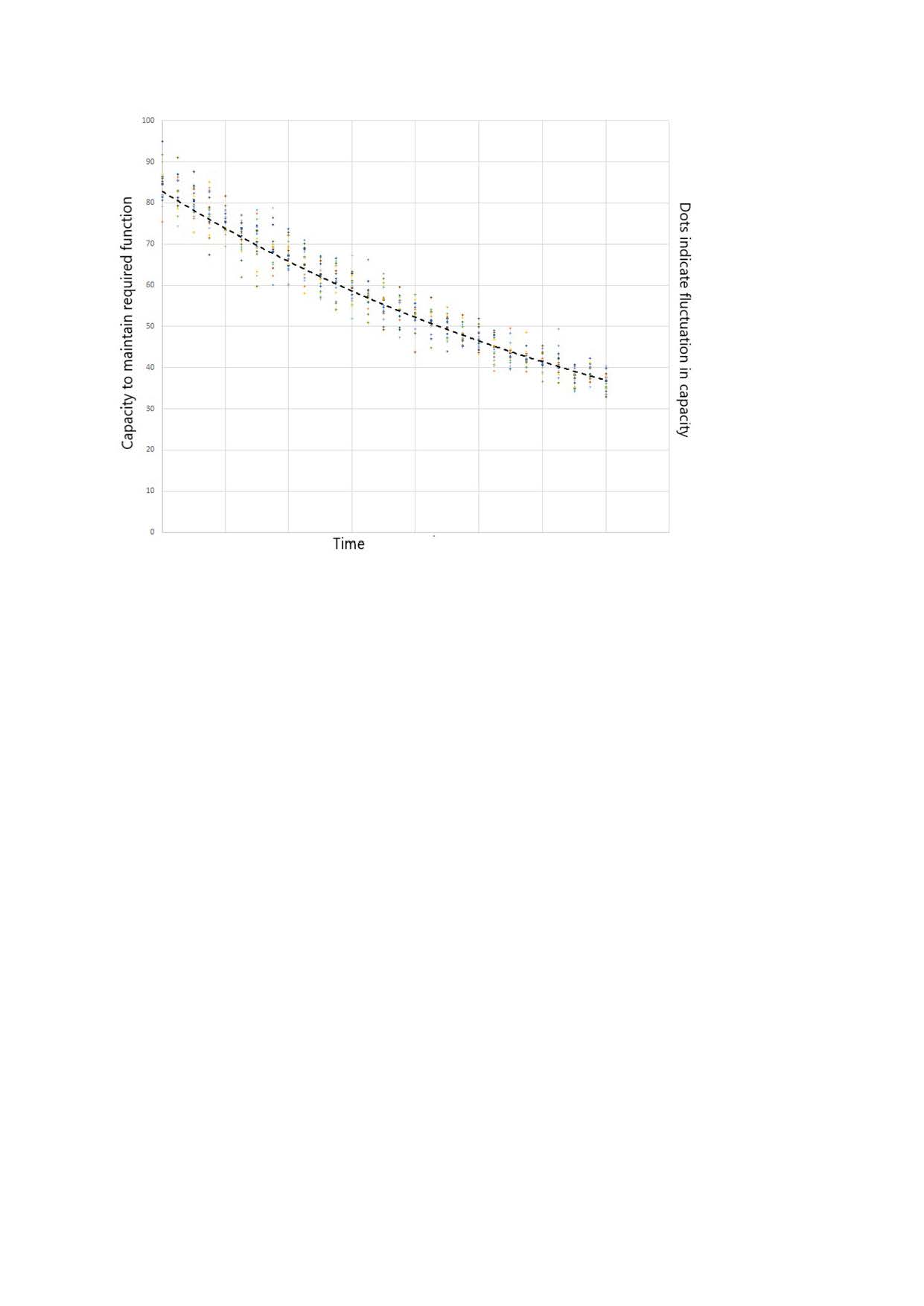
An exponential decay trajectory is followed.
Changes in restorative sleep, supply of energy and supply of oxygen for example cause variation in capacity. Indicated as dots on the graph, with a bell curve distribution about the dashed black curve.
A threshold level below which full function is not possible can then be shown on the graph.
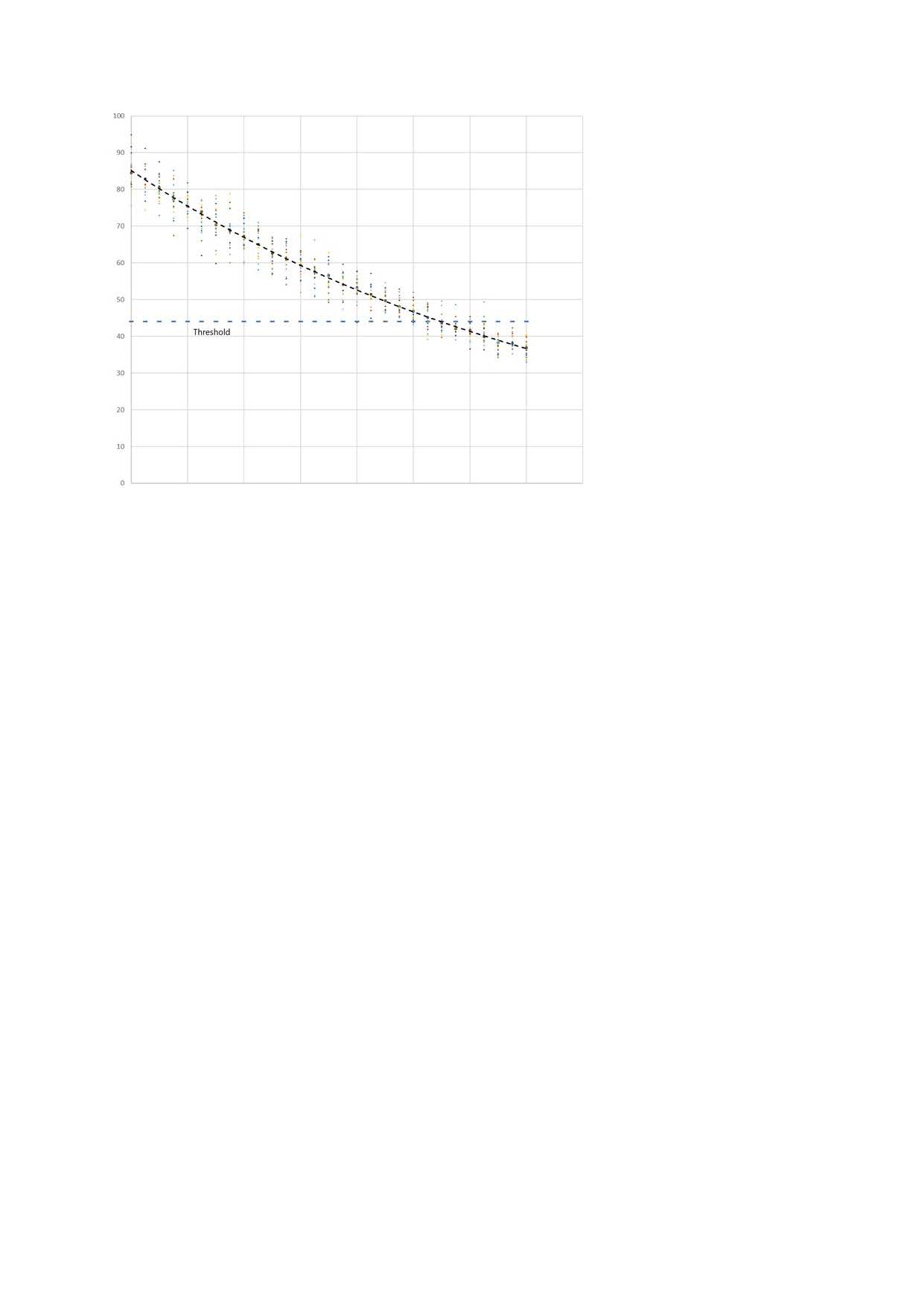
Even before the threshold is reached permanently there will be some periods when variation will cause exceedance. Function will be impaired.
These periods occur more frequently as the threshold is approached, and will last for longer.
The time variation of the probability of threshold exceedance is shown in the following graph.
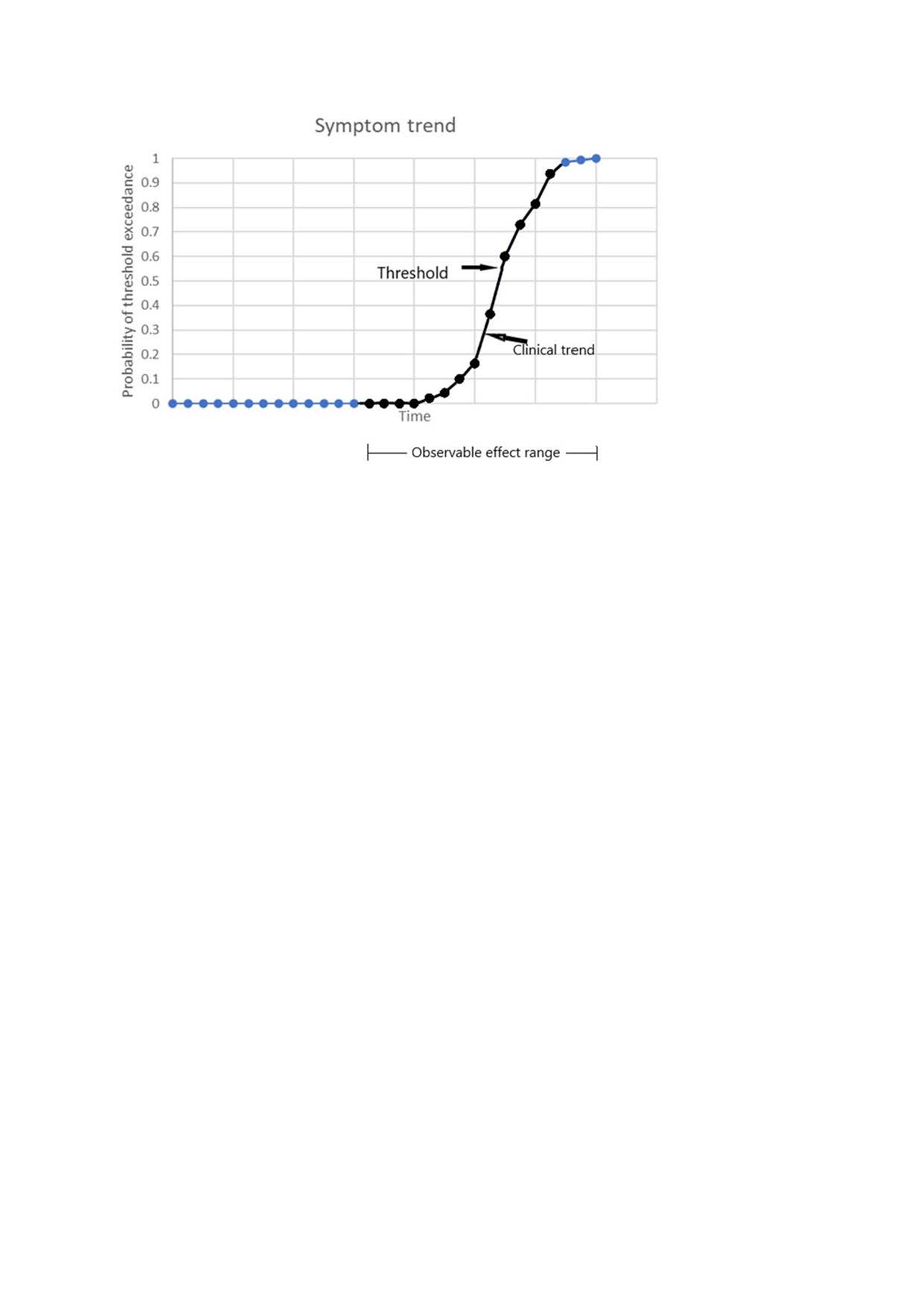
At first any changes in symptoms are very unlikely. As the threshold is approached there is an exponential acceleration of symptom frequency (indicated in black). After threshold, the symptoms trend towards permanence.
A steady exponential decay in neurone number, in combination with fluctuating performance and a function threshold easily explains the apparently logistic clinical trend.
There is no accelerated neurone death and no need to invoke an “infection” model.
So…
Each of the drivers for an “infection” model has been shown to be a result of misapprehension. Even the often observed accelerated frequency of symptoms has a robust explanation based on facts.
For medico-legal purposes the conclusions are that dementia is an advancing disease, not a progressive one. Dementia is divisible, not indivisible.
Can the relationship between TBI and dementia be explained?
The most useful study of this phenomenon corrected for a wide range of risk factors and resulted in a graph of risk vs time between TBI and dementia diagnosis, as follows:
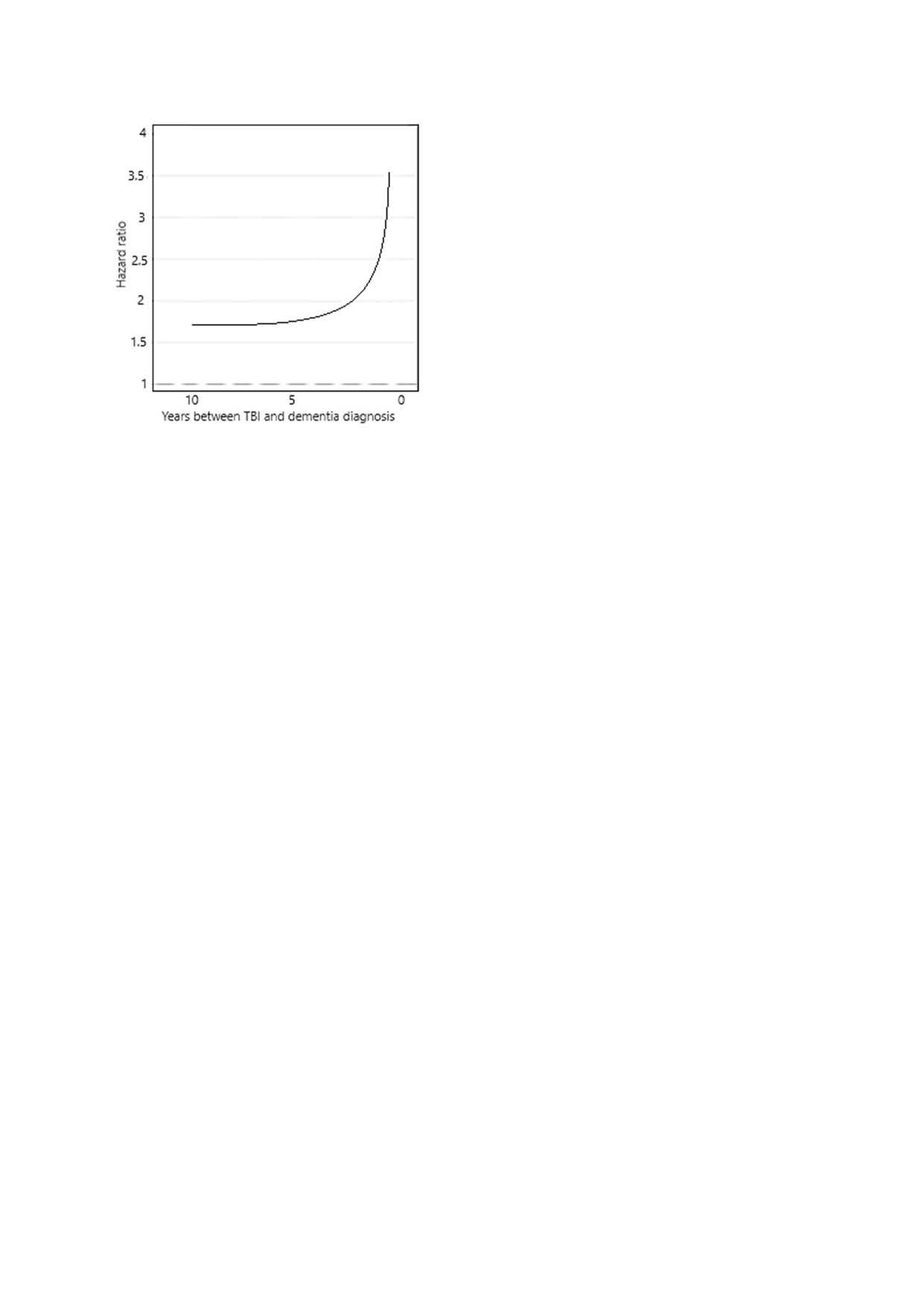
On the left is a clear indication that for those with a TBI there is a raised risk of dementia detection. Or equally, for those with dementia there is a raised risk of a history of TBI. The mathematics cannot distinguish between these two alternatives.
The graph is horizontal at first with a hazard ratio greater than 1.0. that is, there is a real association between dementia and TBI.
If the TBI is the reason for increased risk of dementia, years later, then a progressive disease, or “infection” mechanism, cannot give rise to a horizontal relationship. Such a mechanism must at all times show an increasing risk after trigger.
If the TBI resulted in a function threshold shift this would not vary in time.
The graph then increases very quickly at 5 years prior to diagnosis. This is compatible with an infection model but then you would have to argue that at greater than 5 years the infection began and then its effects were removed but at less than 5 years there was no remission. This is implausible.
The alternative is that, as the function threshold is approached, the daily fluctuations in function, e.g. due to a period of disturbed sleep, led to conditions where a severe accident was more likely. The closer to threshold, the greater the risk of an accident. A so-called reverse causation argument. The curve is very similar to (but not exactly the same as) that which would be expected for a gradual approach to threshold, as shown above.
People diagnosed with dementia don’t drive, but if they did the probability of accidents would tend towards an accident every time.
In my view the reverse causation argument is as yet the more robust.
These findings also mean that if TBI really does increase the risk of dementia, or even directly cause it, this cannot be proved in any study where the time between dementia and TBI is less than 7.5 years. At less than this, the study is dominated by reverse causation.
Good studies would also correct for education and childhood disadvantage and all other known risk factors for detecting dementia. Without such correction, studies are only useful for hypothesis generation and should not be interpreted beyond that. Most of the studies where the authors declare that TBI is a cause of dementia have a very short time window (<2 years) and fail to correct for the obvious dementia detection risk factors. They prove nothing, but keep the hypothesis alive and are dutifully counted by science search robots.
What about soccer?
The recent studies of Scottish professional footballers used a middle class control group. The correct control group would have been of the same childhood socioeconomic status as the footballers. Out-field players were found to have a raised risk of dementia.
Head impacts were not measured or recorded in any way. Despite this, the authors portray heading the ball as the likely cause of the detected association. Would that association have been found if the correct control group had been used? The detected association was of the same strength as is generally attributable to childhood deprivation.
Other studies show that concussion in football is related to contacts with the ground, elbows, knees and goal posts and not to heading the ball.
In short, because the obvious risk factors were not corrected for, and positively the wrong control group was adopted, the study was uninterpretable. Sadly, the study has been hailed as proof that headers cause dementia.
Liability?
There is no sound basis for the theory that negligent brain injury or even concussion could trigger dementia.
Liability could be for negligent damage to neurone numbers or for changing the functional threshold or both. In either case the effect is to accelerate the detection of dementia and in neither case is dementia caused or made worse. Dementia is divisible.
The recent case of Mathieu v Aviva [Case No: QB-2018-004679] included a claim for provisional damages. Mathieu was in his 20s at the time. The argument was that TBI could cause dementia in about 40 years hence. The claimant lost because it was unclear that any detected dementia would be readily attributable to the TBI. The claim therefore failed the tests of the discretion to make a provisional award. This is not the same as answering the general causation question.
The claimant expert offered many of the arguments now known to be wrong and yet the defendant expert was not robust in his counterarguments. Perhaps because it was obvious that the discretion test would be decisive.
In full causation trials it is essential that neither side accepts in any way that dementia is mechanistically indivisible or progressive or accepts any of the false premises that support such a view. A detailed critique of the science may help ensure that no defendant is caught off guard and no claimant is persuaded to make hopeful but unsupportable claims.
Summary
Dementia is caused by the loss of brain function resulting from neurone death. It manifests early in people with faster rates of neurodegeneration and lower resilience. That is, some brains get old sooner than others.
Early manifestation IS NOT proof of some specific disease mechanism.
Increased symptom frequency as the disease develops IS NOT proof of an acceleration in neurodegeneration.
Successive effects in different brain parts IS NOT proof of the spread of neurodegeneration from one part to another.
Association with TBI IS NOT proof that TBI causes dementia, most likely the reverse is the case. Developing dementia increases the risk of TBI.
The reported association with soccer is most likely a result of poor study design.
The facts and the concept of human agency are most consistent with brain function as a mostly integrative phenomenon, with adaptive resilience and with a threshold for loss of function. The expert community is or has been until recently, hugely invested in the “infection” model of dementia. The risk now is that the model and its false assumptions are unconsciously propagated by both claimant and defendant experts.